

Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol . 2017 Dec 19;14(1):7-18.
【Key points】
- 多くの原発性免疫不全症候群における免疫制御異常は自己免疫疾患の発現につながる
- 様々な遺伝子変異は、自己免疫だけでなく免疫不全を引き起こす可能性がある
- これらの遺伝子変化とそのpathophysiological consequences病態生理学的な結果を具体的に知ることで、新たな治療法の開発が可能になる
- 原発性免疫不全症候群に関する知識は、リウマチ性疾患の治療にDMARDsを使用する際に起こりうる感染症関連の有害事象の理解を深めることができる
【背景】
-
リウマチ性炎症性疾患や自己免疫疾患の多くは、その発症メカニズムについて仮説は弱いものが多い
-
しかし数年、遺伝学的な研究が進み、多くのリウマチ性疾患で見られる免疫異常と関連するいくつかの遺伝子変異が検出されるようになった
-
自己免疫疾患と原発性免疫不全症の間には、遺伝的な関連や原因について高い重複があり、フランスNational Primary Immunodeficiencies Registry(CEREDIH)の2017年の解析では、生涯を通じて、原発性免疫不全症の患者の26.2%に一つ以上の自己免疫症状または炎症の兆候が観察された
- autoimmune cytopenia自己免疫性血球減少症のリスクは、原発性免疫不全症患者では一般集団の少なくとも120倍高い
このレビューの目的
- 原発性免疫不全症候群の遺伝学的及び病態生理学的基盤についての説明
- またその臨床的な特徴を説明すること
-
臨床的特徴を知ることで、自己免疫と免疫不全の関係についての知識に加えて鑑別診断に有用である。
-
またリウマチ性疾患の治療では選択的生物学的製剤やキナーゼ阻害剤を使用することで、患者の免疫力が相対的に低下した状態になることがある
-
この2つの疾患群の関係が解明されれば、感染症リスクの増加など、これらの治療法の副作用がより理解されるようになるかもしれない
-
本総説では、すべての原発性免疫不全症候群を網羅することはできないので、リウマチ性疾患と最も関連性の高いものに焦点を当てた
-
ゲノムワイド関連研究(GWAS)ではRAとSLEがよく研究されている。
-
100の非MHCリスク遺伝子座に377の候補遺伝子が同定
-
これらの遺伝子のうち98個がRA発症リスクの2倍増と関連
-
そのうち15個は以前に原発性免疫不全症候群と関連した遺伝子と同一である。
-
この15個がコードする分子は以下の通り
-
caspase-8, caspase-10, autoimmune regulator (AIRE), IL-2 receptor α (also known as CD25), receptortype tyrosine-protein phosphatase C (also known as CD45), VDJ recombination-activating protein 1 (RAG1), RAG2, CD40, serine-protein kinase ATM, non-receptor tyrosine-protein kinase TYK2 (TYK2), uracil-DNA glycosylase, IFNγ receptor 2 and interferon regulatory factor 8
-
-
-
-
-
ほとんどのリウマチ性疾患に関連するpolygenic traits多因子形質とは対照的に、monogenic defects単一遺伝子の欠損による自己免疫性疾患があり、以下のものがある
-
下記のperiodic fever syndromes周期性発熱症候群はすべて関節炎を伴うことがある
-
また単一遺伝子の欠損であるため、将来的にはより多くのリウマチ性疾患が明確に定義された遺伝性疾患に分類されるかもしれない
-
familial Mediterranean fever 家族性地中海熱
-
hyper-IgD with periodic fever syndrome 周期熱症候群を伴う高IgD
-
TNF receptor-associated periodic syn dromeTNF受容体関連周期熱症候群
-
Ideficiencies of IL-1 receptor antagonistsL-1受容体拮抗薬の欠損
-
cryopyrin-associated periodic syndromesクリオピリン関連周期熱症候群(cryopyrinopathiesクリオピリン症ともいう)
-
-
RAもSLEもMHC領域の内外にあるいくつかの遺伝子の変異と関連している
-
これらの変異の中には、補体C1q、C2、C4をコードする遺伝子の変異や、GWASによって同定された50の非MHCリスク遺伝子のリスク遺伝子座が含まれる
-
下記の遺伝子群にはいわゆるinterferonopathiesインターフェロン障害の原因となるIFNαシグナル伝達経路の機能獲得(GOF)につながる変異がある。
-
TLR7, TLR8, TLR9, IRF3, IRF5, IRF7, TYK2, STAT4 and IRAK1
-
-
これらの遺伝子の変異は、IFNα産生の増加の要因になっており、SLE患者では、I型インターフェロン(IFNαなど)の産生がRNAや一本鎖DNAによって誘導されるため、このような典型的な特徴が見られる
-
これらの核酸分子を含む免疫複合体もまた、I型インターフェロンの産生を刺激し、自己免疫を促進することができる
-
RNAやDNAの増加は、TREX1、RNASEH2A、SAMHD1、IFIH1、MAVSにおける遺伝子欠損(LOF)変異の結果であり、I型インターフェロンの産生と自己免疫疾患を引き起こす
【リンパ球の発生とトレランスLymphocyte development and tolerance】
-
B細胞やT細胞の発生には、免疫グロブリンやT細胞受容体(TCR)遺伝子の再配列や組み換えが必要であり、後者は胸腺で行われる
-
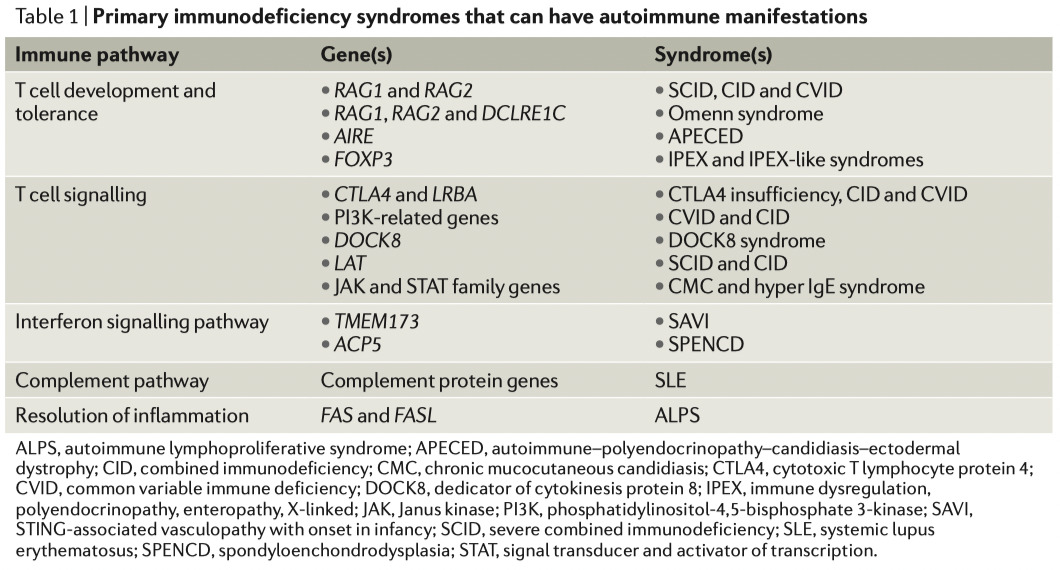
リコンビナーゼ遺伝子であるRAG1とRAG2は、VDJ組み換えで重要であり、他の酵素やリコンビナーゼとともに複合体を形成しVDJ組み換えの際にDNA切断を開始し、その後DNAの切断を修復する
-
その結果、RAGの欠損はsevere combined immunodeficiency(SCID)重症複合型免疫不全症を引き起こし、患者はT細胞やB細胞欠損となり、natural killer cellsナチュラルキラー細胞を持たない状態になる
-
そしてSCID患者は特に日和見感染症に罹患しやすい
RAG低形成変異
-
RAG1およびRAG2における様々なhypomorphic mutations低形成変異の報告がある
-
低形成変異により、residual enzyme activity不完全な酵素活性ができ、そのlevelに応じて、患者はT細胞およびB細胞のレパートリーにおいて異なるlevelのoligoclonalityを表すため、正常なT細胞やB細胞の数は少ないことがある。
-
またRAG1 および RAG2 のhypomorphic mutations低形成変異を持つ患者は、サイトメガロウイルス、Epstein-Barr ウイルス、John Cunningham ウイルスなどのウイルスに慢性的に感染しやすい
-
T細胞やB細胞減少により以下の疾患を引き起こす。
-
idiopathic CD4 lymphopenia 特発性CD4リンパ球減少症
-
common variable immunodeficiency (CVID)分類不能型免疫不全症
-
combined immunodeficiency (CID) 複合免疫不全症(CID)
-
IgA deficiency IgA欠損症
-
Omenn syndrome Omenn症候群
-
RAG低形成変異はOmenn症候群では最もよくある原因であり、この疾患は以下の症状を呈する。
-
lymphadenopathyリンパ節腫脹
-
hepato splenomegaly肝脾腫
-
eosinophilia好酸球増多
-
多臓器におけるオリゴクローナルおよび活性化T細胞の浸潤
-
IgE値の上昇を伴う重度のhypogammaglobulinaemia低ガンマグロブリン血症
-
-
Omenn症候群の症状は、移植片対宿主病GVDHの症状と同様に、autoreactive T cells自己反応性T細胞によって引き起こされる
-
VDJ組み換えに関与するもう一つのタンパク質であるartemisをコードするDCLRE1Cの変異も、Omenn症候群を引き起こすことがある
-
-
-
自己抗体の頻度の増加やB細胞リンパ球増殖の障害、また自己反応性B細胞の選択過程における妨害も、RAG欠損症の患者において観察されている
-
このような患者で見られる幅広い血清中の自己抗体は、alopecia脱毛症、vitiligo白斑、granulomas肉芽腫、myasthenia gravis重症筋無力症、vasculitis血管炎、psoriasis乾癬などの免疫症状と一致している
-
診断においては、phenotypic analysis of lymphocytesリンパ球の表現型分析 、TCR excision circles (TRECs) や κ-deleting recombination excision circles (KRECs)のPCR解析は、遺伝子配列解析の前に使用すれば、T 細胞および B 細胞の発生における異常なスクリーニング方法となる
-
RAG2 欠損症に対する遺伝子治療の前臨床試験の結果は有望であったが、造血幹細胞移植(HSCT) は現在も RAG 欠損症に対する最も成功した治療法である
【Central tolerance中枢性トレランス】
DiGeorge症候群 や autoimmune polyendocrinopathycandidiasis–ectodermal dystrophy (APECED)自己免疫性多発性内分泌疾患-外胚葉性ジストロフィーの2つの疾患は胸腺T細胞の発達障害を特徴とする
DiGeorge症候群
-
染色体22q11.2にヘテロ接合性の欠失があり、the third and fourth pharyngeal pouches第三咽頭袋と第四咽頭袋の不完全な発達を引き起こし、副甲状腺機能低下症、心臓やfacial malformations顔の奇形といった症状を引き起こす
-
Partial DiGeorge syndrome 部分的DiGeorge症候群
-
出生数4,000人に1人の割合で存在し、胸腺が低形成または非典型的な場所に位置している
-
部分的DiGeorge症候群の患者の8.5 – 10%に自己免疫症候群(自己免疫性甲状腺機能低下症、RA、vitiligo白斑、乾癬、autoimmune cytopenias自己免疫性細胞減少症)を認める。これは自己反応性細胞の欠失によるため?である
-
-
免疫不全はT細胞の数と機能に依存しており、観察される表現型は、SCIDのようなT細胞の欠如から、ほぼ正常なT細胞の補完まで様々である
-
T細胞の表現型の重症度によって、感染症に対する感受性のレベルが異なることがある
-
治療法は、ほとんどがexperimental実験的なもの
-
造血幹細胞移植HSCTは、重症の SCID 様表現型の患者には成功率が低いが、胸腺組織移植は、胸腺欠損のある DiGeorge 症候群の患者の治療に成功している
APECED
-
中枢性免疫トレランスを制御する転写因子であるAIREの様々な変異によって引き起こされるmonogenic disorder単一遺伝子疾患である
-
これらの患者では、胸腺上皮細胞での AIRE の欠損によって T 細胞の選択が障害を受けるため、制御性 T 細胞(Treg) の発達にknock-on effectノックオン効果(波及効果)をもたらし、T reg 細胞の減少を引き起こしてしまう
-
APECEDの患者は通常、内分泌器官における自己免疫症状を呈し、副甲状腺、副腎、生殖腺、甲状腺に影響を及ぼす
-
最も深刻なものは患者の90%以上が罹患する副甲状腺機能低下症と副腎機能不全
-
患者の 80%以上に慢性粘膜皮膚カンジダ症を認める
-
患者の大多数が、IL-17、IL-22、IFNω26 , 27 に対する自己抗体を持っている
-
治療は通常、ホルモン補充と必要な場合の免疫抑制である
【Peripheral tolerance末梢性トレランス】
-
T reg 細胞によって制御されており、表面抗原 CD3、CD4、CD25 の発現と、転写因子 FOXP3 の発現によって特徴づけられる
-
complex autoimmune syndromes患者におけるMonogenic mutations単一遺伝子突然変異はヒトの免疫恒常性の維持、特に腸管におけるTreg細胞の重要性を証明している
-
ヒトでは、T reg細胞の重要性の発見は、X染色体上にあるFOXP3の突然変異の同定より見いだされ、FOXP3の変異はimmune dysregulation免疫調節障害、polyendocrinopathy多発性内分泌障害、enteropathy腸疾患、X-linked (IPEX) syndromeX連鎖(IPEX)症候群に関連する
-
これらの患者は下痢をし、早期に発症する臓器特異的自己免疫により、内分泌腺(膵臓や腸を含む)などの臓器が破壊
-
マウスのFOXP3変異はIPEX(X-linked) に類似した症状を示す「scurfy」表現型を引き起こす
-
IPEX syndrome患者ではFOXP3変異はT reg細胞の欠如を引き起こすのみであるが、これはこの多臓器免疫疾患を引き起こすのに十分である
-
マウスでのAdoptive T cell transfer experimentsによって、Treg細胞の欠如が特に腸の免疫恒常性に影響を与えることが示されている
-
-
IPEX様症候群の患者におけるFOXP3遺伝子座のエピジェネティック解析により、FOXP3の変異が明らかでない場合でも、T reg細胞の数が減少すると、自己免疫疾患につながる可能性があることが示された
-
この研究では、患者の半数が敗血症、サイトメガロウイルス、上気道感染症、肺炎などの感染症を発症していた
-
-
Treg細胞は、自己免疫を防ぐために組織に存在する必要があるだけでなく、正確に機能する必要がある
Treg細胞とIL-2 受容体 (CD25) の関連
-
ある患者群では、T reg細胞の機能障害が、CD25の欠損によることが判明しており、IL-2 受容体 (CD25) は、 T reg 細胞が正しく機能するために特に重要であることがわかっている
-
自己免疫疾患の予防として、治療的に T reg 細胞数を回復させる試みがあり、またSLE 患者において、 低用量 IL-2 療法は T reg 細胞数の増加に有効であり、 他の自己免疫疾患にも応用できる可能性がある
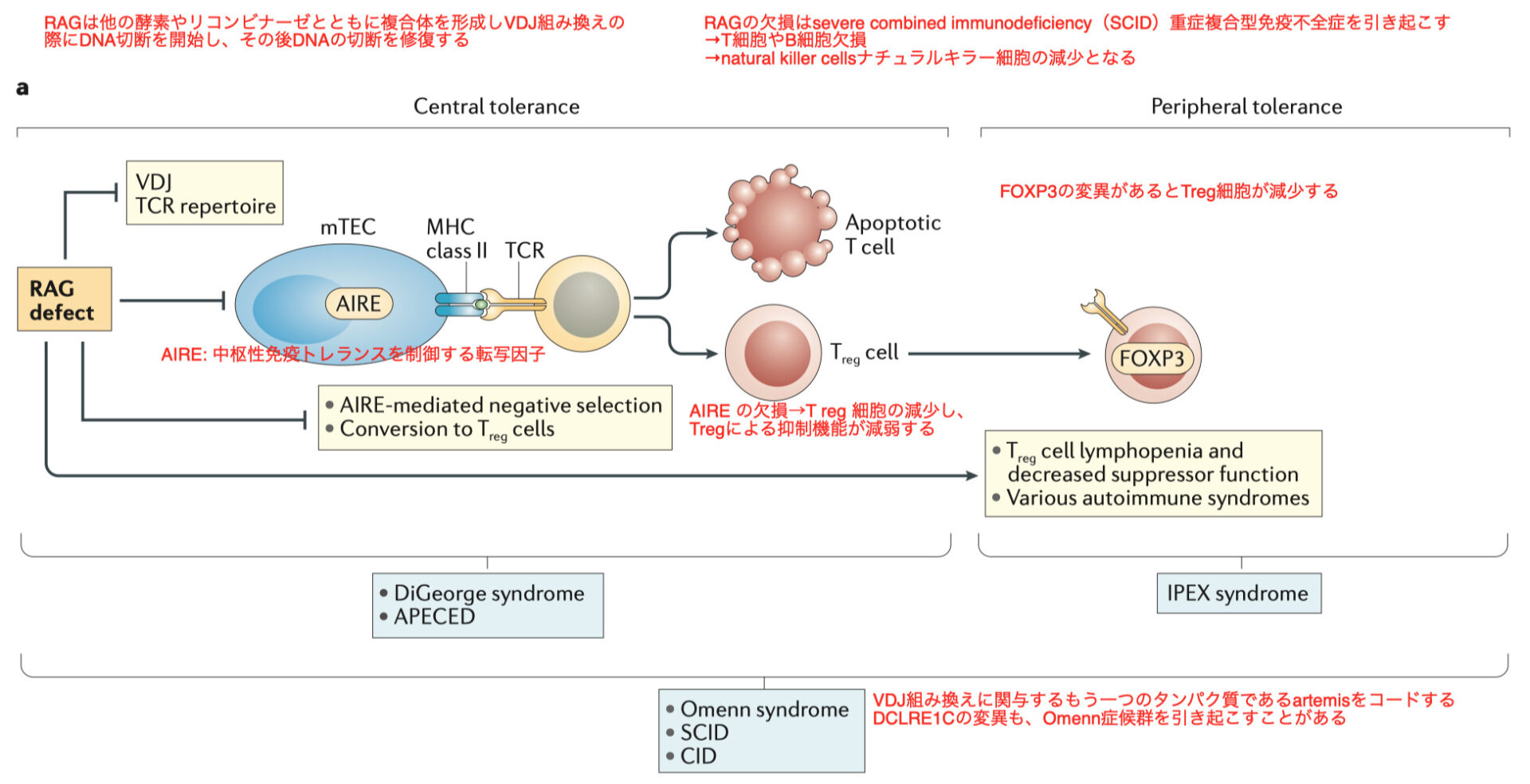
補足:Tregの発生と機能について (アバスの基礎免疫学第6版より
CD4+T細胞→(転写因子FoxP3の影響)→T reg細胞に分化
T reg細胞はCD4陽性でIL2Rのα鎖であるCD25を高発現しており、FoxP3を発現している
IL-2は活性化T細胞から作られ、活性化T細胞の増殖を誘導して免疫応答を増強する一方でT reg細胞の機能を維持して免疫応答を抑制する。相反する作用をもっている
- ナイーブT細胞の活性化の抑制
- エフェクターT細胞への分化の抑制
- NK細胞やB細胞の抑制
※IL2Rの高頻度に発現し、IL-2を消費して活性化T細胞の増殖を抑制する経路もある
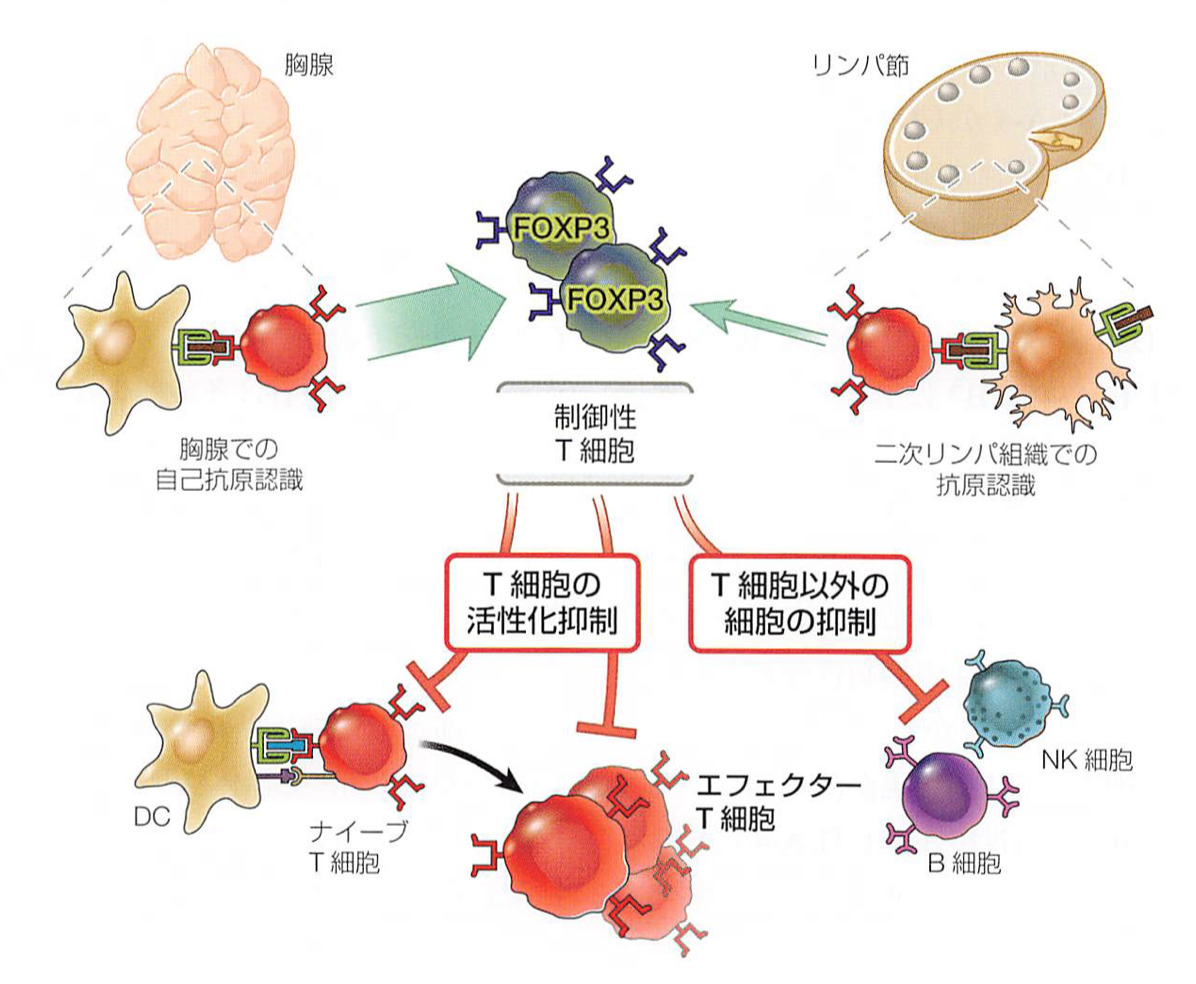
【T細胞シグナル伝達 T cell signalling】
-
T細胞は2つの重要なシグナルによって活性化される
-
第1の刺激シグナルは、HLA分子上に提示された抗原とTCRが結合すること
-
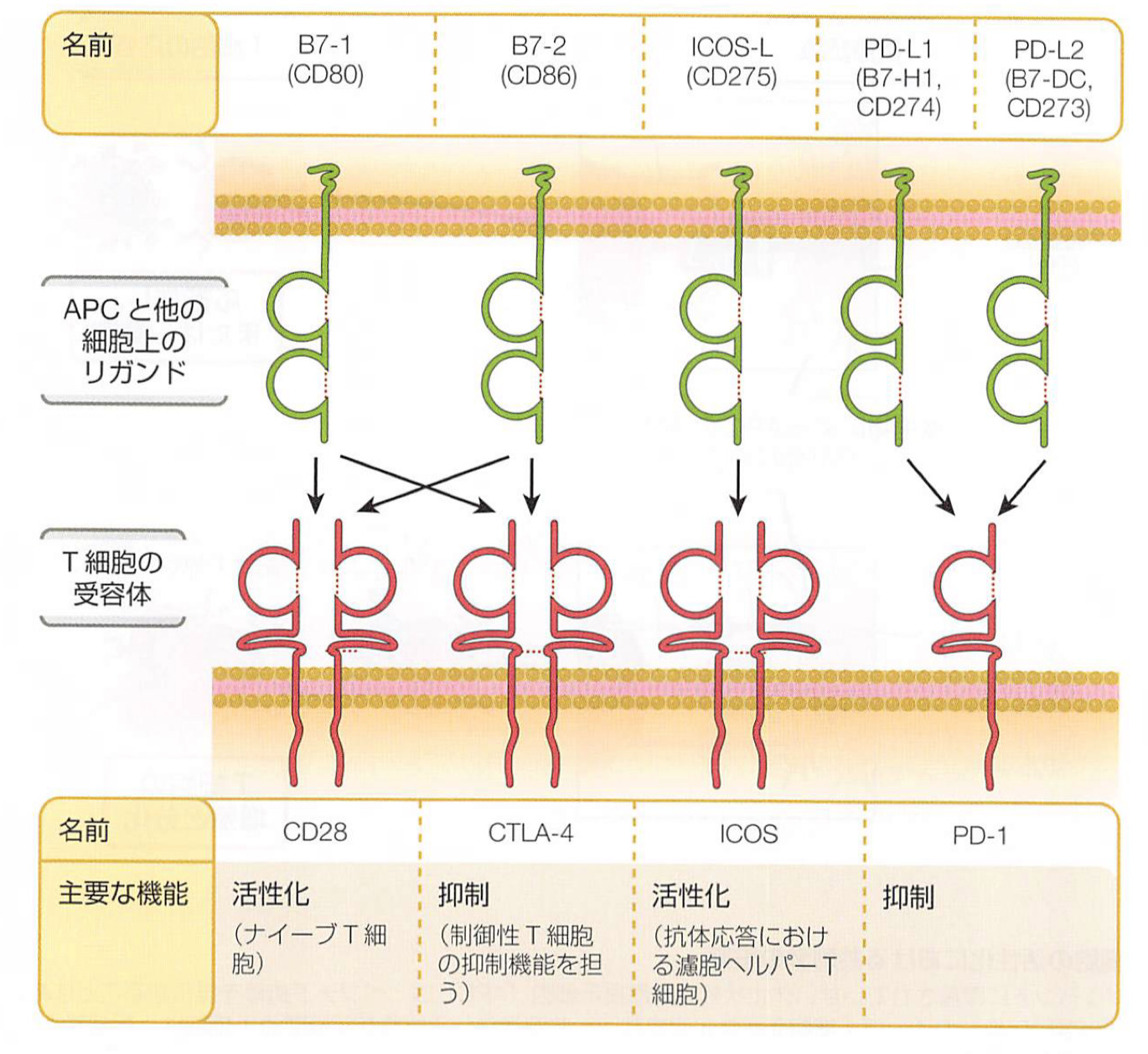
第2のシグナル(共刺激シグナルとして知られている)は、共刺激分子CD28および誘導性T細胞共刺激因子(ICOS)によるもの
-
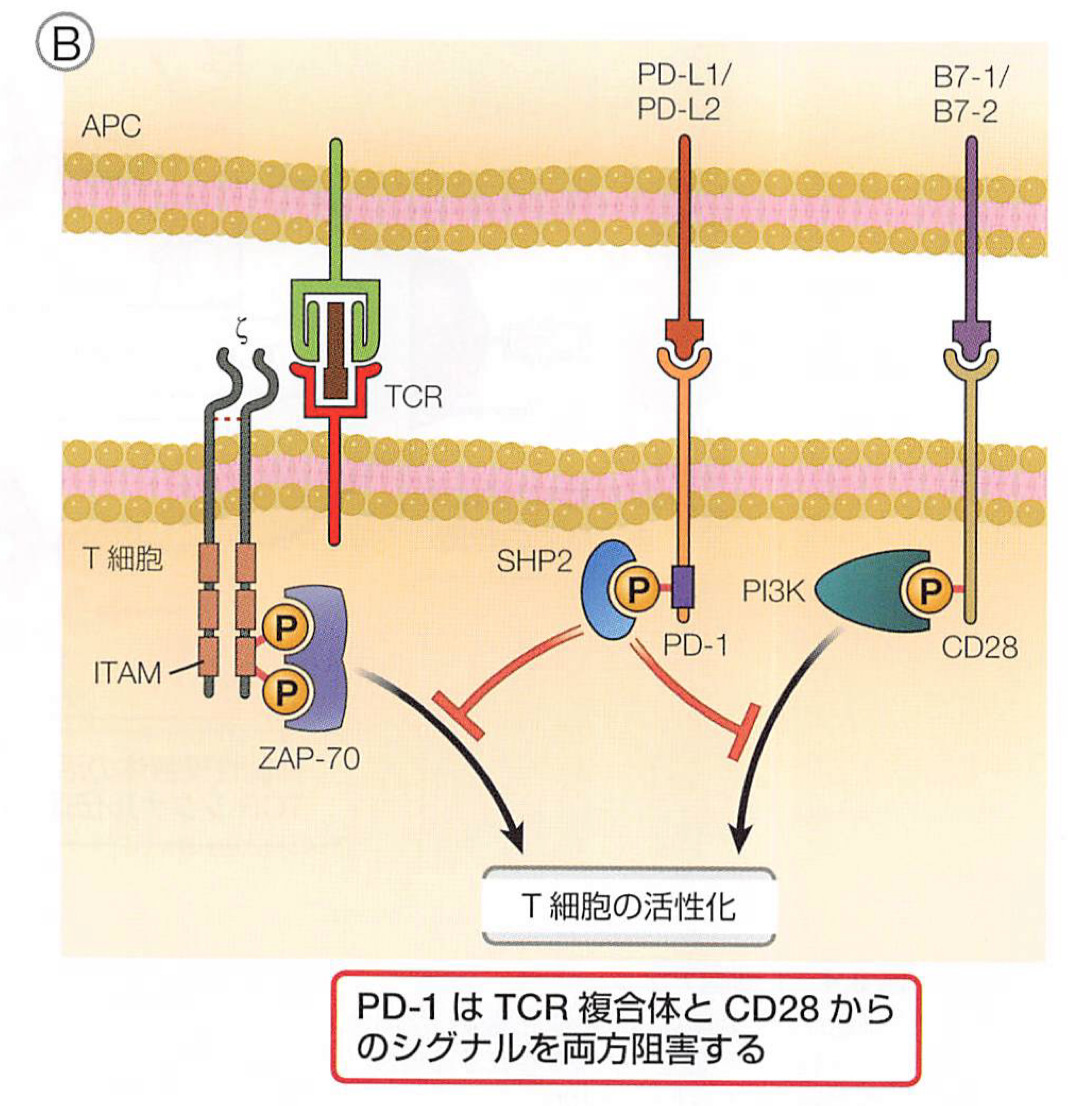
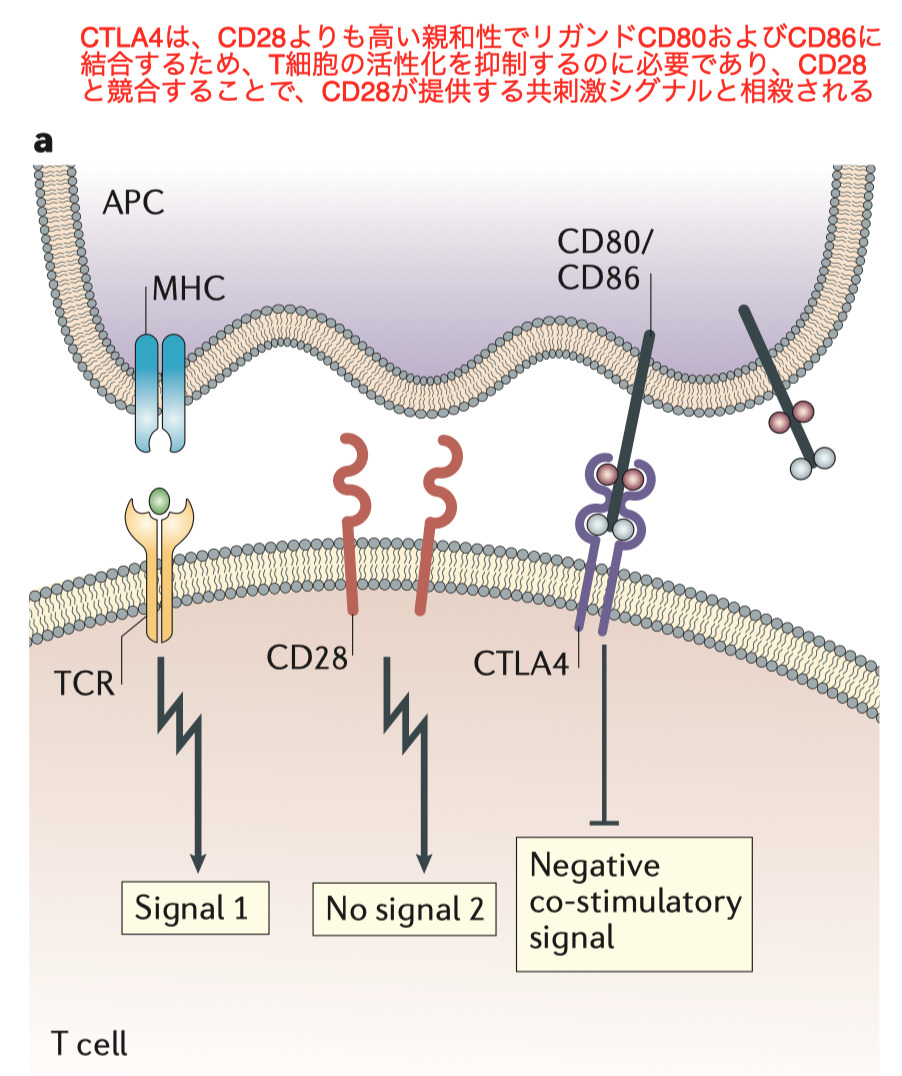
共刺激はcytotoxic T lymphocyte protein 4 (CTLA4)とprogrammed cell death protein 1 (PD1)の2つの膜貫通型受容体によって細かく調節され、これらは細胞の活性化によりT細胞表面で発現が増加する
-
これらの抑制性のレセプターがそれぞれのリガンドに結合することで、T細胞の活性化が抑制される
-
このような堅実に制御されたメカニズムが阻害されると、T細胞の恒常性が不安定になり、免疫介在性疾患につながる可能性がある。
-
T細胞活性化の亢進をもたらすperturbation摂動は自己免疫を引き起こす
-
一方でT細胞アナジーにつながる摂動は、再発性かつ重度の日和見感染症や腫瘍の発生につながる素因となる
補足:自己抗原を認識するとき、 T 細胞は CD28 ファミリーの共刺激因子群のなかの CTLA-4 (CD l52 ) や PD-I (CD279) などの抑制性受容体を優先的に用いる。アナジーT細胞はこれらの抑制性受容体を高発現している
※CTLA4・・・T 細胞の抑制性受容体であり制御性 T 細胞のエフェクター 分子でT1DやRAに関与
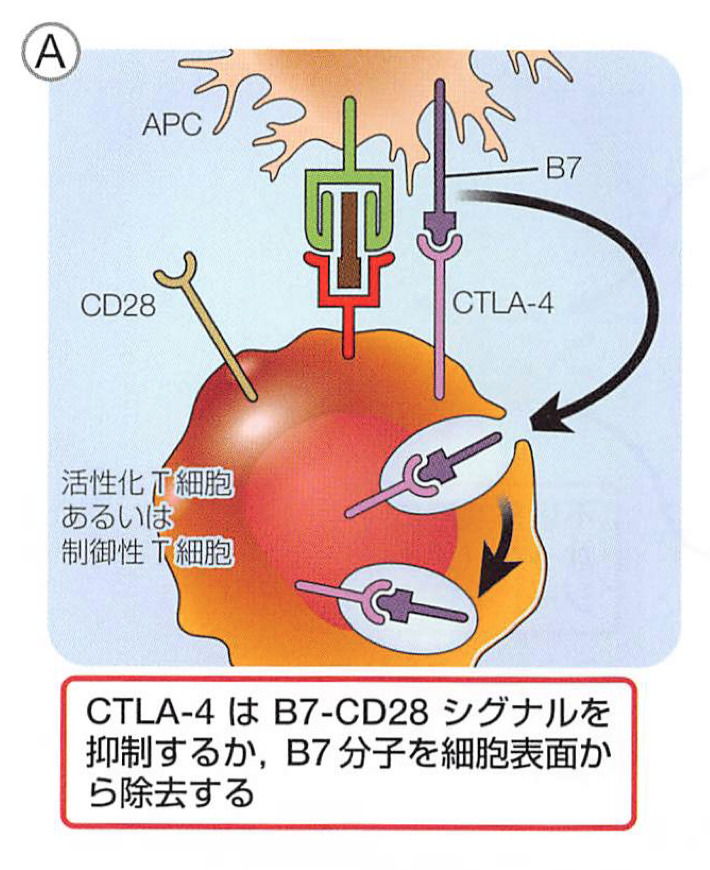

【免疫調節の異常 Abnormal immune regulation】
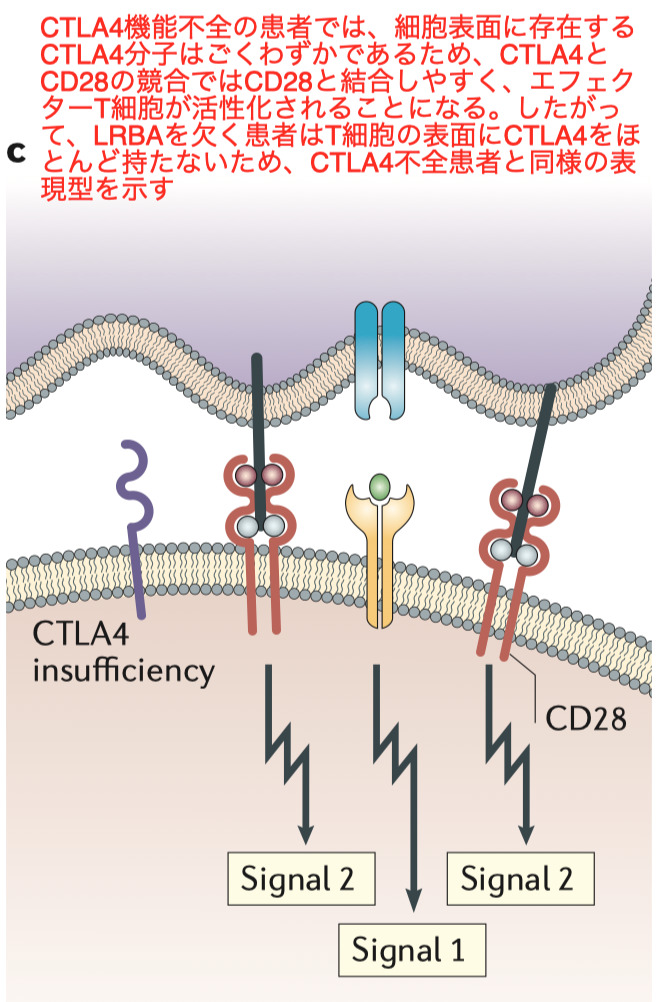
-
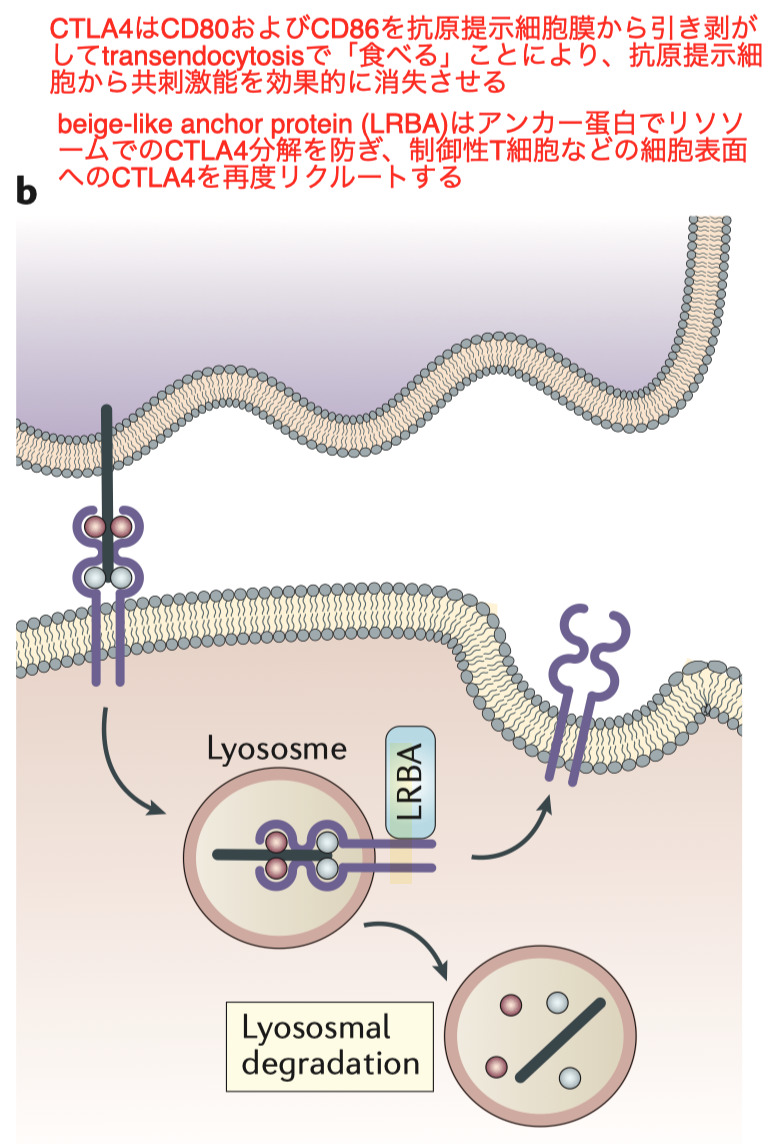
primary immunodeficiency syndromes原発性免疫不全症候群でみられるCTLA4 機能不全と LRBA 欠損の表現型は、 活性化したエフェクター T 細胞による臓器への浸潤とautoimmune cytopenias自己免疫性血球減少症の発生によって特徴付けられる
-
CTLA4 signallingは2つのCTLA4-Fc融合タンパク質であるabataceptとbelataceptによって効果的に利用されており、それぞれRAと腎移植後のT細胞媒介移植片拒絶反応の治療に適応がある
-
両分子とも、CTLA4欠損症およびLRBA欠損症に対する臨床試験を実施されている
-
SLE やその他のリウマチ性疾患におけるヒドロキシクロロキンの治療効果も、CTLA4 発現への影響に起因すると考えられている
【異常な免疫活性化】
-
原発性免疫不全症候群の研究により、T細胞の活性化を引き起こすいくつかの単一遺伝子の欠陥が同定された
-
PIK3CD・・・白血球に選択的に発現するタンパク質であるphosphatidylinositol 4,5-bisphosphate 3-kinase subunit δ(PI3Kδ)をコード
-
→リンパ球におけるPI3Kδシグナルが増加
-
→RACαセリン・スレオニン・プロテインキナーゼ(AKT;プロテインキナーゼBとも呼ばれる)のリン酸化が亢進
-
→mTOR(mechanistic target of rapamycin)の活性化やリボソーム蛋白S6キナーゼβ1のリン酸化が亢進
-
→T細胞の増殖やエフェクター機能の発達をおこる
-
-
現在、PIK3CDの活性化変異を持つ患者に使用するPI3Kδ阻害剤が検討されている
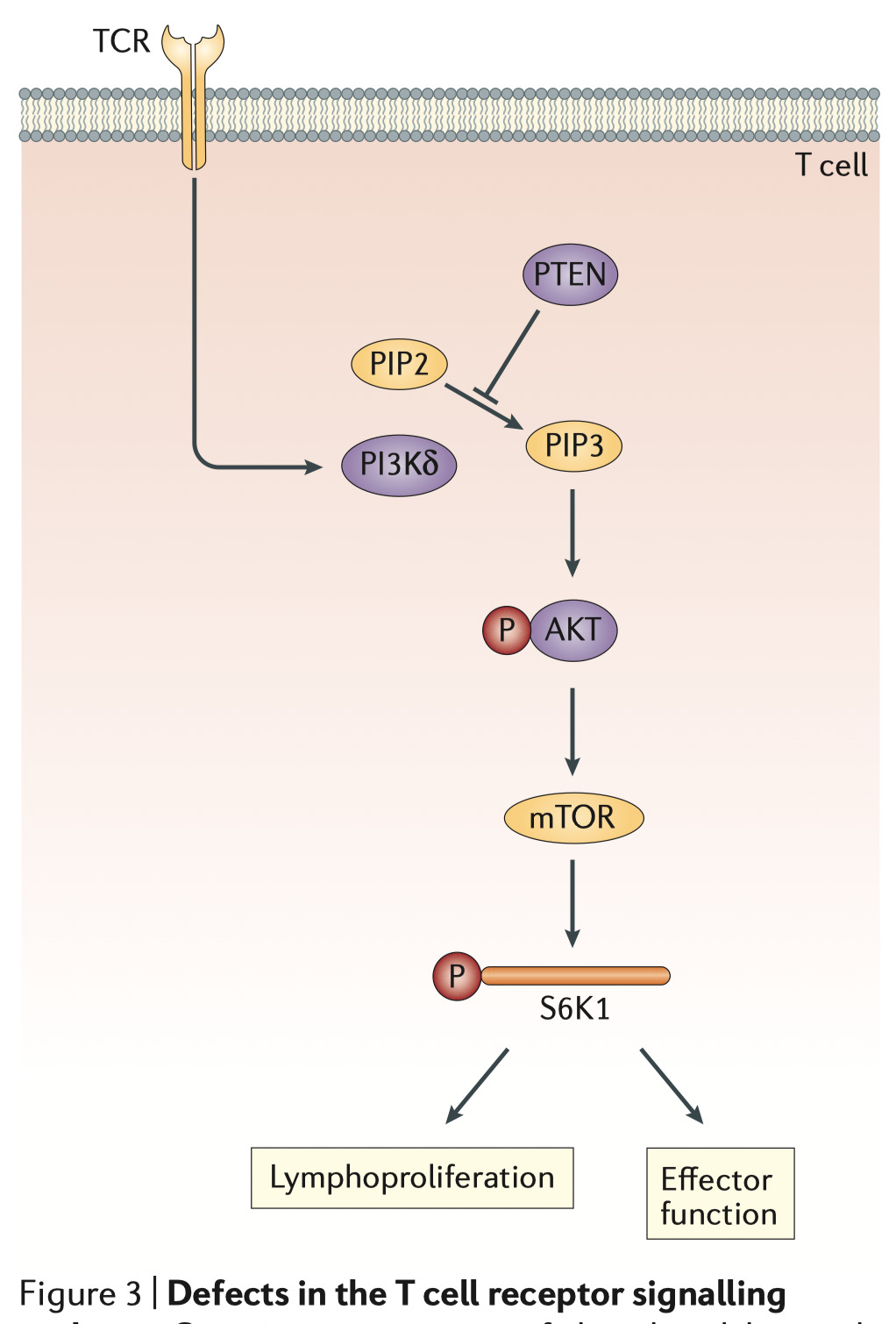
-
このようにT細胞の活性化が構成的に増加する患者は全身性の自己免疫 を発症し、腸、肺、造血系(autoimmune cytopenia)に最もよく影響を与える
-
罹患した臓器には通常、顕著なT細胞浸潤が見られる、また臓器全体を破壊し、臓器不全や死に至ることがある
-
しかし、すべての患者が同じ表現型を示すわけではなく、特に常染色体優性型の疾患では、浸透率の低下と発現率の大きな変動が関与している
-
第二の遺伝子変異やエピジェネティックな変化、環境要因(例えば、感染症)が、このような自己免疫の浸透度や発現率に影響を与えるかどうかは、現在研究中である
-
これらの単一遺伝子疾患は、T細胞の過活性化が自己免疫につながり、選択的mTOR阻害剤(sirolimusシロリムスなど)や共刺激シグナルの阻害が、自己免疫疾患の理想的なtargeted treatmentsとなり得ることを説明している
【TCRのシグナル伝達TCR signalling】
-
2009年に常染色体劣性遺伝のdedicator of cytokinesis protein 8(DOCK8)欠損症のCID患者が報告された
-
DOCK8はアクチン細胞骨格の制御やシグナル伝達物質・転写活性化物質3(STAT3)の活性化における免疫機能における中心的な役割があある
-
DOCK8の欠損により以下のプロセスの機能不全がおこり、感染症の再発やアレルギー性疾患(湿疹やアレルギーなど)、自己免疫、ウイルス性の悪性腫瘍が生じる
-
cell polarization and migration 細胞極性化や遊走
-
cell adhesion 細胞接着
-
immune synapse formation 免疫シナプス形成
-
regulation of STAT3 STAT3の制御
-
phosphorylation and translocation of STAT3 to the nucleus STAT3のリン酸化や核への移行
-
cytolytic granule release 細胞溶解による顆粒の放出
-
actin cytoskeleton organization and dysfunction of T reg cell suppressive function アクチン細胞骨格構成およびT細胞抑制機能の障害
-
- SCID患者や自己免疫症状を持つcombined immunodeficiency (CID) 複合免疫不全症患者では、linker for activation of T cells (LAT)T細胞活性化リンカー(LAT)の変異が報告されており、exson 5 のホモ接合型変異、 premature stop codon早期の停止コドン、 完全な LOF に至るタンパク質の切断を引き起こす
- CIDの3人の患者は早期に自己免疫症状を呈していた。リンパ球数および抗体価は当初正常であったが、病気の進行に伴い、リンパ球減少や日和見感染症を認めた
(LATとはアダプタータンパク質で細胞内のシグナル伝達経路に関与 するタンパク質でT 細胞の活性化に関与する。他にSLP-76やGrb-2 などがある)
【JAK-STATシグナル経路JAK–STAT signalling pathway】
- 1989年に初めてJanus kinases (JAKs)ヤヌスキナーゼが報告された
- 様々なサイトカイン(type I IFNs, type II IFNs, granulocyte-macrophage colony-stimulating factor, several interleukins, erythropoietin, growth hormone and prolactin)がそれぞれの受容体に結合すると、JAKはリン酸化され、次に転写因子のSTATファミリーをリン酸化する
- JAKがリン酸化されると、そのSH2ドメインを介して特定のSTATをリクルートし、それがJAKの基質となる
- リン酸化されたSTATは受容体から遊離し、二量体化して核に移動し、そこで特定のenhancer elementsエンハンサーに結合して遺伝子の転写を開始させる
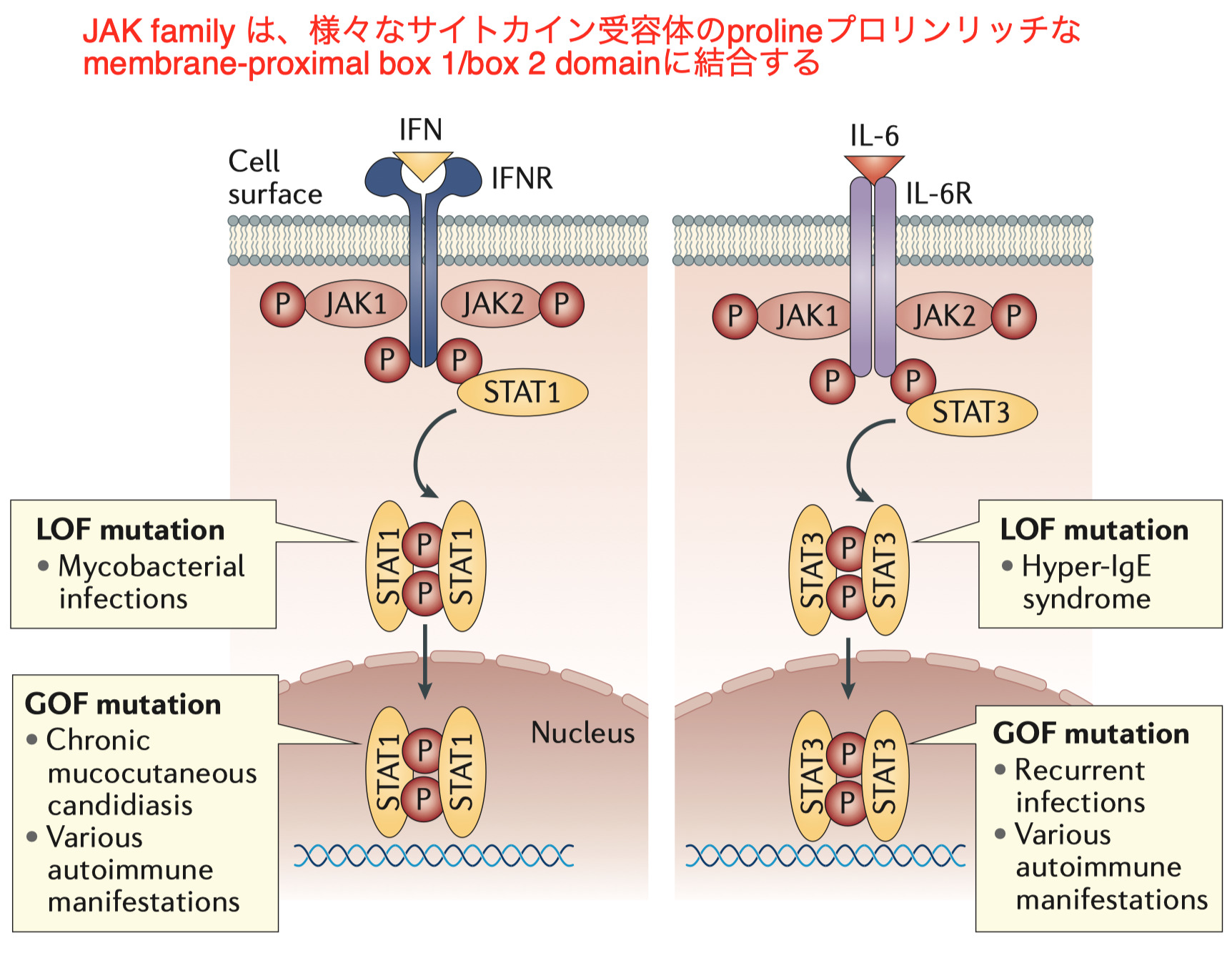
-
哺乳類のSTATファミリーには、STAT1、STAT2、STAT3、STAT4、STAT5a、STAT5bおよびSTAT6の7つがあり、特定のJAKは特定のSTATに優先的に結合するが、ほとんどのサイトカインは複数のSTATを活性化する
-
多くの遺伝子は複数のSTAT分子によって制御されているため、異なるサイトカインの遺伝子活性化プロファイルには、多くの重複とpleiotropy多相遺伝が存在する
JAK分子やSTAT分子の変異について
STATの変異は、STATシグナルを減少させるもの(LOF変異)と、STATシグナルを増加させるもの(GOF変異)のいずれか
-
JAK1のGOF変異はatopic dermatitisアトピー性皮膚炎、eosinophila好酸球、hepatosplenomegaly肝脾腫、autoimmune thyroid disease自己免疫性甲状腺疾患を引き起こす
-
JAK2 の GOF 変異 (Val617Phe) は真性多血症などの骨髄増殖性疾患を引き起こす
-
JAK2 がサイトカインのシグナル伝達経路に関与するだけでなく、 エリスロポエチンやトロンボポエチンのシグナル伝達経路に関与していることが原因であると考えられている
-
-
JAK3の二重性変異は、T細胞における重要なサイトカインシグナル伝達の欠如により、SCIDを引き起こす
-
STAT1 GOF 変異はヘテロ接合体であり、STAT3 シグナリングを相対的に減少させることにより、T ヘルパー17 (TH 17) 細胞の発生を阻害する
-
TH 17細胞の欠如は、IL-17とIL-22の欠如につながり、これらは特に皮膚や粘膜において、Candida属やブドウ球菌に対する骨髄由来のdanger signals危険信号の増幅に重要な2つのサイトカインである
-
ゆえに、STAT1 GOF 変異を持つ患者は、慢性粘膜皮膚カンジダ症にかかりやすく、しばしば毛嚢炎や皮膚膿瘍を発症する
-
-
これらのGOF遺伝子変異を有する患者ではIFNαシグナルは増加する
-
甲状腺機能低下症、自己免疫性肝炎、I型糖尿病、idiopathic thrombocytopenic purpura特発性血小板減少性紫斑病、autoimmune haemolytic anaemia自己免疫性溶血性貧血などの自己免疫疾患を引き起こす
-
-
STAT1 LOF 変異による常染色体劣性遺伝性疾患では、TH 1 細胞の分化が阻害され、I 型および II 型 IFN 産生が低下する
-
STAT1 LOF 変異を持つ患者は、mycobacterialマイコバクテリアやウイルスに感染しやすいという特徴を持つが、heterozygousヘテロ接合型の STAT1 LOF 変異では、anti-mycobacterial defence system抗マイコバクテリア防御システムのみが損なわれている
-
-
STAT3 GOF 変異は、indolent lymphoma低悪性度リンパ腫に分類されるlarge granular lymphocytosisと呼ばれるリンパ増殖性障害を引き起こす
-
Heterozygous germlineヘテロ接合性の生殖細胞系列 のSTAT3 変異も観察され、リンパ増殖と自己免疫血球減少症、肺・胃・腸・肝臓の病変・多関節炎などの早期発症の自己免疫症状と関連している
-
-
STAT3 LOF 変異(STAT3 dominant-negative mutationsとも呼ばれる)は、高 IgE 症候群(別名 Job 症候群または Buckley 症候群)と呼ばれる表現型を引き起こし、これは再発性のskin boils皮膚腫脹、肺炎および高濃度の IgE を特徴とする
-
STAT1 GOF 変異と同様にSTAT3 LOF 変異もTh 17 細胞不足を引き起こし、患 者はブドウ球菌やカンジダ属菌に感染しやすくなる
-
-
JAK阻害剤をリウマチ性疾患患者の治療に使用する際はこれらのJAK及びSTAT分子のLOF変異とウイルス感染との関連を念頭に置く必要がある
-
遺伝子変異とは異なり、JAK 阻害剤はサイトカインシグナルを完全に遮断しないため、studyで観察された感染症の発生率は低い
-
しかし、JAK 阻害剤の投与開始後、herpes zoster infection帯状疱疹の感染率が上昇する
-
-
現在、 脊椎関節炎の治療法として IL-17 blockadeが行われているが、 この治療法を受けた患者の感染率は大きくないが、 TH 17 細胞を欠く患者の経験から予測されるように、 カンジダの発症率が高い
【インターフェロンシグナル経路Interferon signalling pathway】
-
内因性のDNAやRNA分子は、エンドソームのToll様受容体(TLR3、TLR7、TLR8、TLR9)、細胞質RIG-I様ヘリカーゼ、特殊なDNAセンサーcGASを介して炎症カスケードを誘発することができる
-
これらの経路はすべて、IFN制御因子の活性化に続いてI型IFNを誘導する
-
interferonopathies IFNの異常は自己免疫や免疫不全を引き起こすが、これはヌクレアーゼの欠陥によってのみ引き起こされるだけではなく、自然免疫センサーやadaptive receptor適応型受容体が、連続して活性化したり感受性が高まったりする状況でも自己免疫に異常がでる
-
I型IFNの負の制御因子の欠損がこのような臨床像を引き起こすことがある
-
I型IFNは、SLEの病態に最も重要なサイトカインである
-
SLEの既知のIFN関連の遺伝的なリスク因子に加えて、多くのIFN異常が発見されている
-
例えば、IFN遺伝子刺激タンパク質(STING)をコードするTMEM173の変異は、STING活性にGOFを与える
-
このような変異は、I型IFNを構成的あるいは環境刺激依存的に誘導し、STING関連血管障害(SAVI)などの疾患を引き起こして全身性炎症、CRP値の上昇、重度の血管障害を伴う
-
-
Spondyloenchondrodysplasia脊椎軟骨異形成症(SPENCD)は、ACP5の低形成変異(ACP5: 破骨細胞分化のマーカーである酒石酸耐性酸性フォスファターゼ5型をコードする)のを伴い、endochondral bone growth軟骨内骨成長の障害とtype I IFN signatureを引き起こすもう一つのinterferonopathy,インターフェロン疾患である
【免疫複合体とアポトーシスの残骸Immune complexes and apoptotic debris】
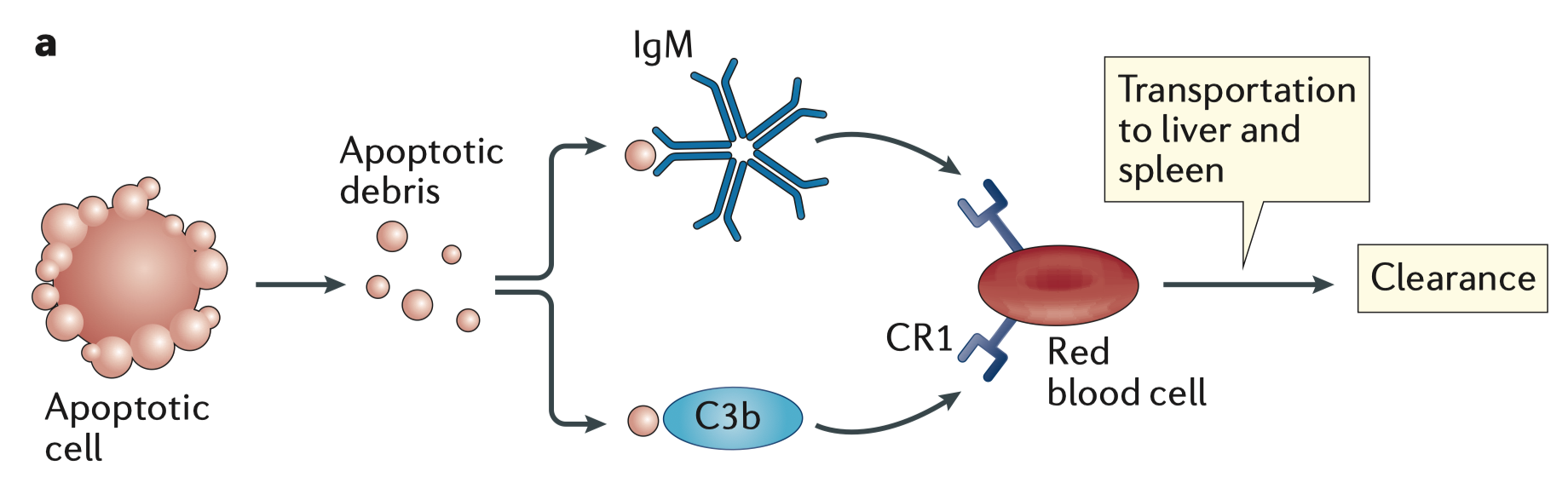
-
免疫複合体の除去は、細胞の残骸が補体タンパク質、自然抗体(主にIgM)や特異的IgG抗体と結合し、その後肝臓や脾臓の細胞によってこれらの複合体が取り込まれることによっておこる
-
→補体タンパク質のうち、免疫複合体は主に補体タンパク質C3bと結合し、赤血球上の補体受容体1と結合する
-
→赤血球は、結合した免疫複合体を肝臓や脾臓に運び、そこで免疫複合体は貪食される
-
補体蛋白質C1q、C1r、C1s、C2、C4などの補体経路のearly part初期部分の成分の不全は、C3bの低レベルと関連しており、免疫複合体のクリアランスの障害の要因となる
-
このような状況では、血液中の細胞性物質が長時間存在すると、自己抗体の形成を誘発する可能性があり、免疫複合体自体が自己免疫反応をさらに刺激する可能性がある
-
Sjögren syndrome-related antigen A (SSA; also known as Ro)シェーグレン症候群関連抗原 A(SSA、別名 Ro)とanti-SSA antibodiesの抗 SSA 抗体の複合体はTLR と Fc 受容体をcrosslinking架橋してマクロファージによる TNF 産生を刺激する
-
U1 small nuclear ribonucleoprotein(U1snRNP)と抗 U1snRNP 抗体(SLE 患者にみられる)の複合体は形質細胞性樹状細胞による IFNαの 産出を誘発する
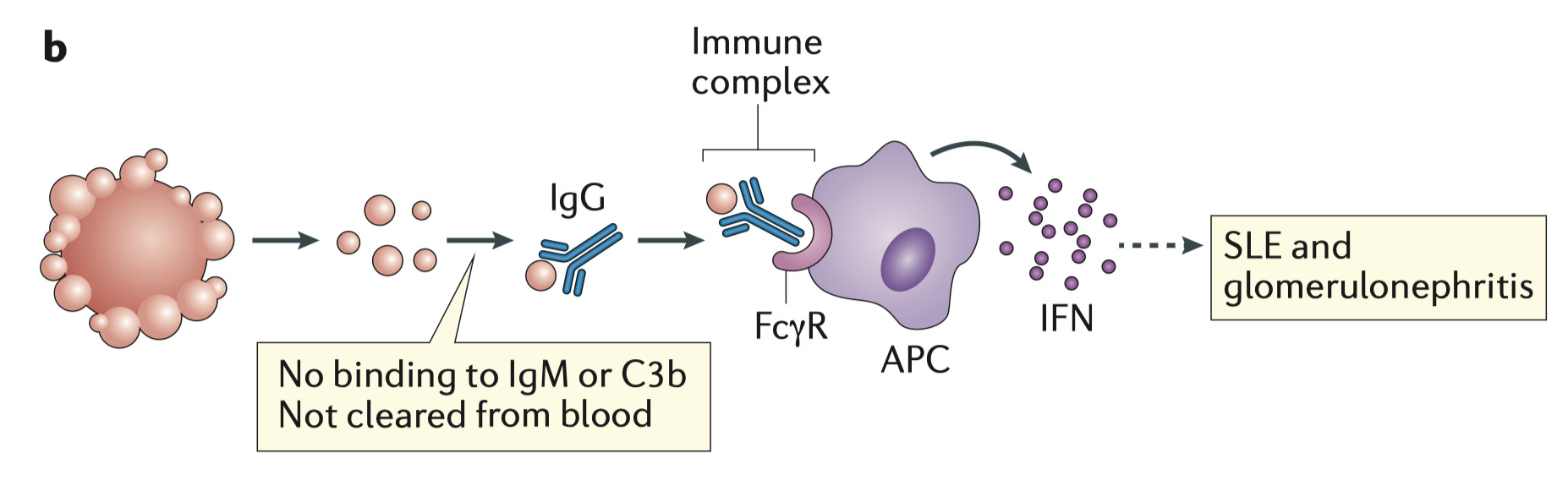
-
complement deficiencies補体欠損症やselective IgM deficiency選択的IgM欠損症の患者では、細胞のdebrisが循環から除去されないかわりに、IgG抗体の産生を刺激する
-
IgGとdebrisの免疫複合体はマクロファージや樹状細胞のFc受容体に結合し、I型IFNや他のサイトカインの産生を誘発する
-
特に、I型IFNの産生が亢進すると、全身性エリテマトーデス(SLE)の発症につながることがある
-
また二本鎖DNA(dsDNA)やヌクレオソームと抗dsDNA抗体との免疫複合体は、糸球体基底膜に沈着するため、SLE患者において糸球体腎炎を誘発しうるかもしれない
-
一方で、natural IgM antibodies自然のIgM抗体はある種の自己抗原によるダメージに対して保護的である可能性がある
-
lupus傾向のあるNZB×NZW F1マウスにIgM抗dsDNA抗体を注射すると、糸球体腎炎の発症を防ぐことができた
-
このモデルマウスでは、IgMとdsDNAの複合体は肝臓で除去されるため、IgGとdsDNAの病原性複合体は形成されない
【補体経路Complement pathway】
-
稀ではあるが、補体経路の初期部分の構成要素の欠損とSLEと強く関連しており、C1またはC4欠損の人の80%、C2欠損の人の30%で発症する
-
C2 deficiency C2欠損症は、ヨーロッパでは1万分の1から2万分の1の割合で発生し、小児のupper respiratory tract infections上気道感染症にも関連するprimary immunodeficiency syndrome原発性免疫不全症候群である
-
完全 C3が欠損すると、幼少期に重篤な細菌感染症(特に肺炎球菌やインフルエンザ)にかかりやすくなる
-
このような初期の感染症から生き延びた子供たちは、その後、糸球体腎炎のような免疫複合体を介した自己免疫症状を発症する
-
注目すべきは、ヘテロ接合性のC3欠損症の場合はC3の血清濃度の低下を伴うが、臨床的に関連した症状は生じない
-
the membrane attack complex膜侵襲複合体(C5C9)の構成要素の欠損は、Neisseria spp.の感染症にかかりやすい
-
しかし、これらの補体タンパク質は免疫複合体のクリアランスに関与しないので、これらの欠損は自己免疫と関連しない
-
-
補体受容体3(CR3;CD11bとしても知られている)は不活性化C3bに結合するインテグリンであるが、その完全な欠損はまれであり、重度の細菌感染症やSLEと関連している
-
CR3 のArg77His 変異は、不活性化 C3b で被覆された赤血球の貪食を阻害し、SLEと関連している
-
IgGを含む免疫複合体に対するphagocytesの反応はそのような免疫複合体とFcγレセプターとの相互作用によって影響を受けている
-
Fcγ受容体3(FcγRIII;CD16)およびFcγRI(CD64)は活性型受容体であり、一方、FcγRIIb(CD32bとしても知られている)は抑制型受容体である
-
FcγRIIbのIle232Thr変異体は、マクロファージやB細胞におけるこの受容体の正常な阻害活性を消失させ、SLEと関連しているが、興味深いことにマラリアからの保護も提供する
-
一方で、FcγRIIIb をコードする FCGR3B にはコピー数の変異が存在し、コピー数が少ないと、phagocytesによる免疫複合体のクリアランスが妨げられ、SLEと関連があるとされている
-
現在、soluble Fcγ receptor constructs可溶性 Fc γ受容体コンストラクトを用いた新しい治療法が研究されており、免疫複合体と Fc 受容体の相互作用を阻害することを目的としている
【炎症の終わりResolution of inflammation】
感染に反応して増殖する免疫系の細胞は、理論的には、免疫系の過剰活性化や自己免疫を避けるために、感染の脅威が去ればその数は減少するはず
T細胞の死には、大きく分けてACADとAICDの2つのメカニズムがある、AICDについて詳細に紹介
-
activated T cell autonomous death (ACAD)活性化T細胞自律死はTCRの再刺激とは無関係に起こる
-
activationinduced cell death (AICD)活性化誘導性細胞死は、主にTCRを介して再刺激を受けた活性化T細胞に起こる
-
AICD は、 IL-2、IL-4、IFNγ やアポトーシス制御因子 BCL2 や BCL2-associated agonist of cell death などの抗アポトーシス機能を持つタンパク質など多くの因子に影響される
-
活性化B細胞やT細胞上のFasや活性化T細胞上のFasリガンド(FasL;TNF ligand superfamily member 6)の発現が増加し、FasとFasLの相互作用によりカスパーゼカスケードを引き起こし、アポトーシスに至る
-
Autoimmune lymphoproliferative syndrome (ALPS)自己免疫性リンパ増殖症候群はアポトーシスの欠陥によって引き起こされすが、主にCD3+ CD4- CD8- T細胞のリンパ球増加と、polyclonal高ガンマグロブリン血症によって特徴づけられる
-
ALPS 患者は脾臓腫大、リンパ節腫脹、そして頻繁に自己免疫性血球減少症や自己免疫性臓器疾患を発症するが、癌、特にリンパ腫の発症リスクも高い
-
ALPS患者の大半はFASにヘテロ接合性の生殖細胞変異または体細胞変異を持ち、少数の患者はFASLGに変異を持つ
-
-
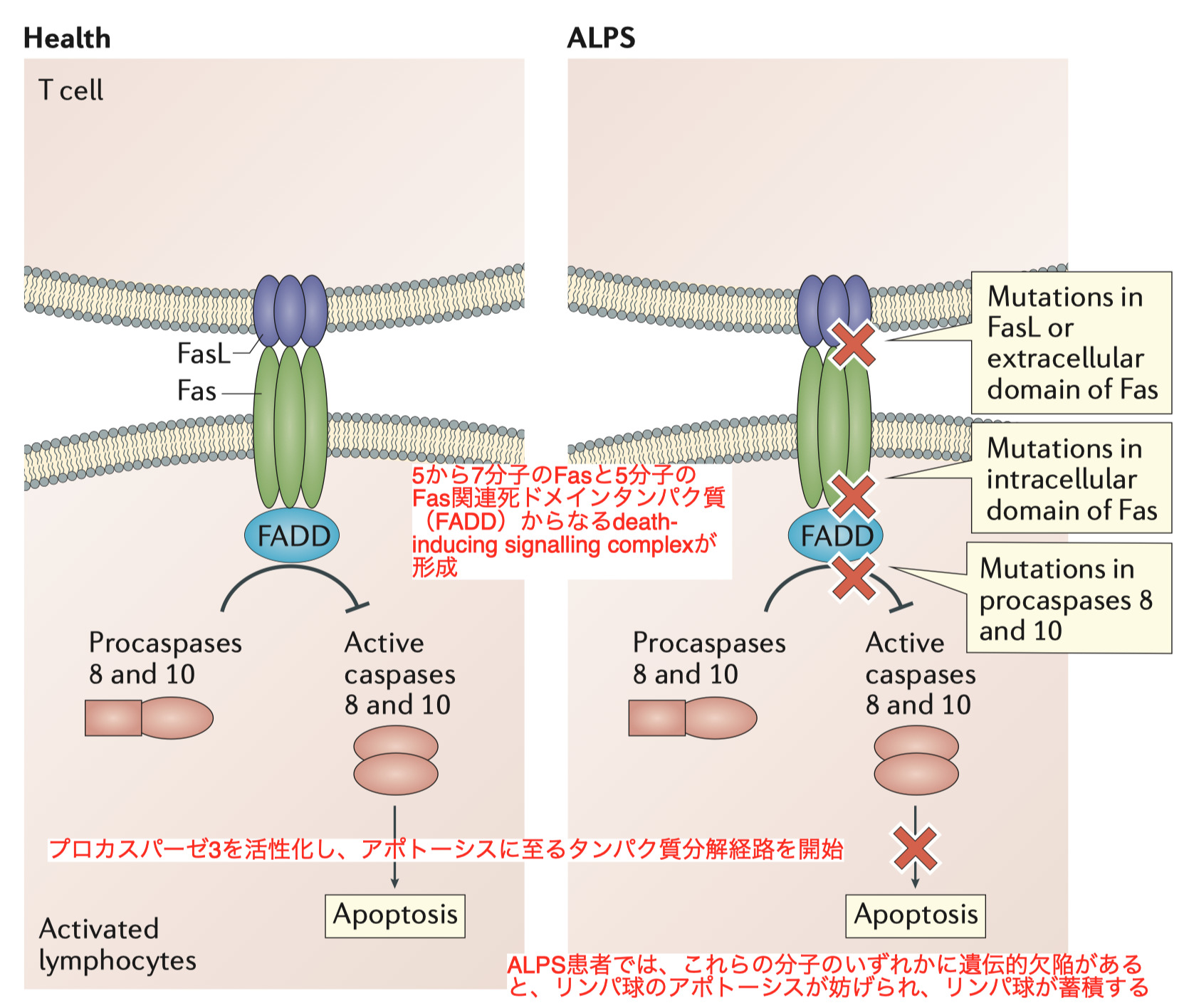
-
Fasの変異を持つlupus-proneループス傾向のMRL/lprマウスは、抗核抗体と抗dsDNA抗体を伴うlupus-like疾患を発症する
-
マウスのリンパ球増殖障害は、アポトーシスに影響を与えるFasの欠陥で説明できる
-
-
FAS変異を持つヒトは通常dsDNAに対して抗体を持たない
-
またmTOR 阻害剤である sirolimus は、ALPS 患者の拡大した CD3+ CD4- CD8 – T 細胞数を正常化して、臨床症状を軽減する効果がある
-
SLEや関節炎、その他の自己免疫疾患といった疾患は、しばしばchronic granulomatous disease慢性肉芽腫性疾患を伴うことがある
-
これらの患者では、NADPH oxidase complexの構成要素の変異により、活性酸素の産生が減少し(通常、phagocytes)によるオートファジーやT細胞の活性化を抑制する)、炎症亢進の状態が引き起こされる
AICD APECED CD25 chronic granulomatous disease CTLA4 DiGeorge症候群 Fas FOXP3 IL-2 IPEX JAK LRBA MAVS RA RAG SCID SLE STAT Treg 原発性免疫不全症候群




